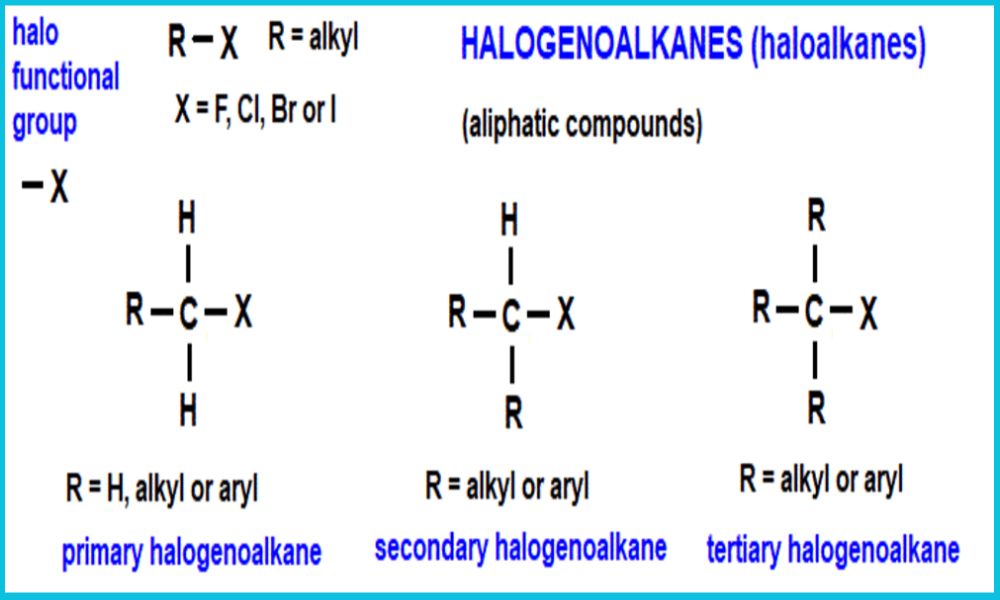
Introduction
Organic chemistry relies heavily on rearrangement reactions for their production and transformation of organic compounds. Hydride shifts and methyl shifts are two common intramolecular rearrangements which often take place, so understanding their differences is vital in understanding organic reactions as a whole and predicting outcomes accurately.
Hydride shifts involve the movement of an H- group from one carbon atom to another within the same molecule, usually during reactions involving carbocation intermediates, to stabilize positive charges and form more stable carbocations compounds. Hydride shifts may have significant ramifications on regioselectivity and stereochemistry of reactions.
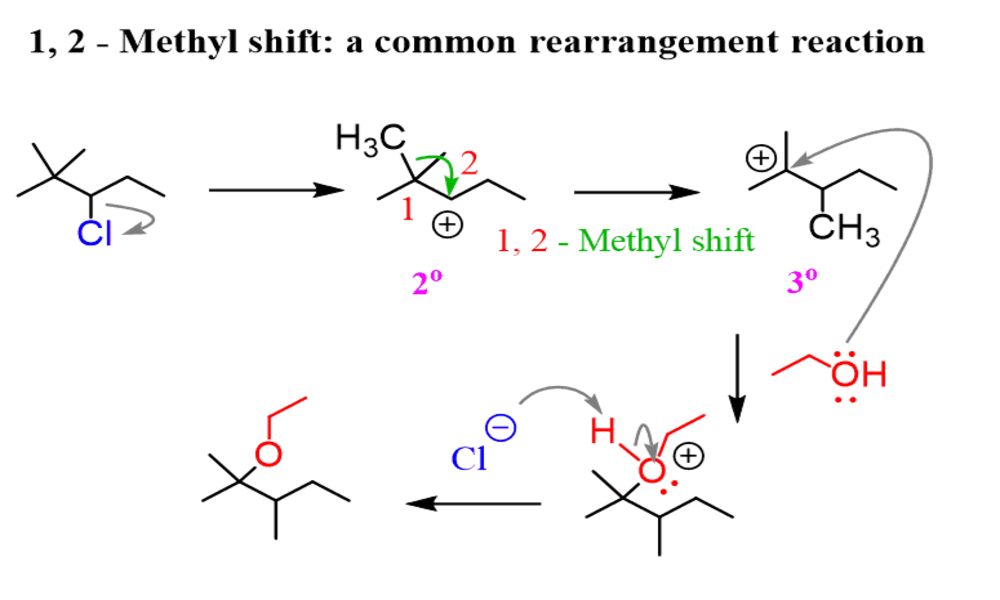
Methyl shifts involve the movement of methyl groups (-CH3) from one carbon atom to another within the molecule, similar to how hydride shifts occur during reactions involving carbocation intermediates; they enable more stable carbocations formation while altering reaction outcomes and can even result in different isomers or regioisomers being formed, often seen in organic reactions.
Distinguishing between hydride and methyl shifts is essential, as they have distinct properties and affect reaction outcomes in different ways. This content outline will explore their definitions, mechanisms, occurrences and examples to better illustrate their significance in organic chemistry and further assist chemists with designing complex organic molecules through increased understanding.
Definition of hydride shift
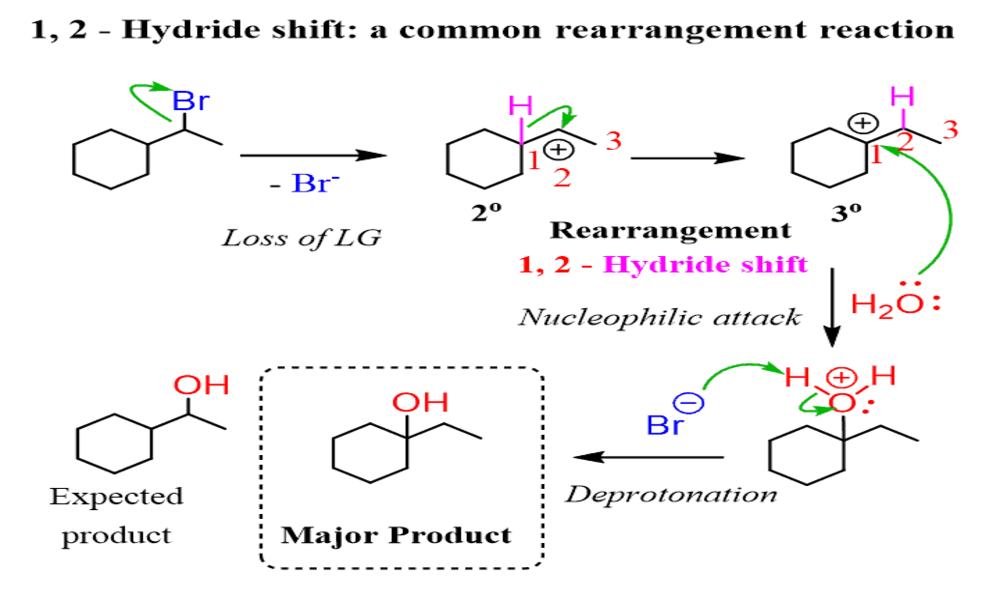
Hydride shift is an intramolecular rearrangement reaction in organic chemistry whereby hydrogen atoms (H-) and their accompanying electrons move from one carbon atom to an adjacent carbon atom or another atom in the same molecule, creating new carbon-carbon or carbon-heteroatom bonds.
Hydride shifts often take place during reactions involving carbocation intermediates, where migration of the hydride group helps stabilize positive charge by creating more stable carbocations intermediates. They play an integral part in various transformations and play key roles in complex molecular transformations and creation processes.
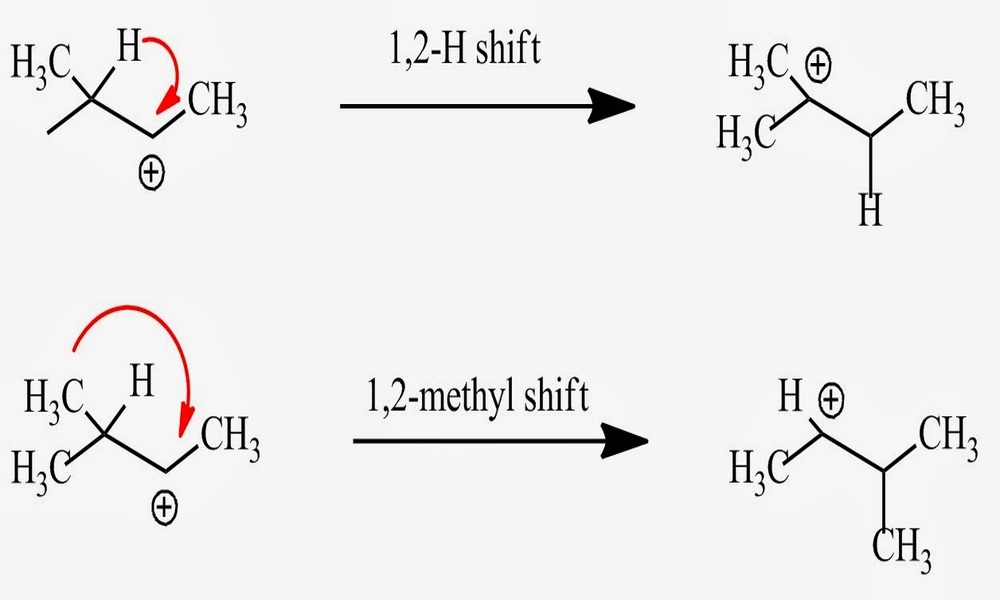
Definition of methyl shift
A methyl shift is an intramolecular rearrangement reaction in organic chemistry in which a methyl group (-CH3) moves from one carbon atom to an adjacent carbon atom or another site within the same molecule. Methyl shifts occur frequently during reactions involving carbocation intermediates, similar to hydride shifts.
Methyl shifts play an essential role in organic synthesis and are often observed during carbocation rearrangements or radical reactions involving carbon. They provide stability by creating more stable carbocations structures. Methyl shifts can result in isomerization of different structural isomers or regioisomers, leading to different isomeric forms or isomer subclasses arising.
Comparison Table of Hydride and Methyl Shift
Here is a comparison table highlighting the key differences between hydride and methyl shifts:
| Accept | Hydride Shift | Methyl Shift |
|---|---|---|
| Definition | Migration of a hydride group (H-) from one carbon atom to another within the same molecule | Migration of a methyl group (-CH3) from one carbon atom to another within the same molecule |
| Group Migration | Hydride group (H-) | Methyl group (-CH3) |
| Reaction Type | Commonly observed in reactions involving carbocation intermediates | Commonly observed in reactions involving carbocation intermediates |
| Stabilization | Stabilizes positive charge on the carbon atom by forming a more stable carbocation | Stabilizes positive charge on the carbon atom by forming a more stable carbocation |
| Regioselectivity | Can influence regioselectivity by changing the position of the migrating hydride group | Can influence regioselectivity by changing the position of the migrating methyl group |
| Stereochemistry | Generally does not affect stereochemistry | Generally does not affect stereochemistry |
| Bond Dissociation Energies | Involves the migration of a relatively low-energy hydride group | Involves the migration of a higher-energy methyl group |
| Intermediates Stability | Results in the formation of more stable carbocation intermediates | Results in the formation of more stable carbocation intermediates |
| Examples | Alkyl rearrangements, carbocation stabilization | Carbocation rearrangements, radical reactions |
By examining this comparison table, we can see that while both hydride and methyl shifts involve the migration of a group within a molecule, there are distinct differences between them. Hydride shifts involve the migration of a hydride group, influencing regioselectivity and stabilizing carbocations.
Methyl shifts, on the other hand, involve the migration of a methyl group, also impacting regioselectivity but generally not affecting stereochemistry. Understanding these differences is crucial for predicting the outcomes of organic reactions and designing synthetic strategies in organic chemistry.
Importance of understanding the difference between hydride and methyl shifts
Understanding the differences between hydride and methyl shifts is of immense value in organic chemistry for various reasons:
Prediction of Reaction Outcome: Recognizing both hydride and methyl shifts allows chemists to more accurately predict the outcomes of organic reactions. By understanding which shift is involved, one can anticipate any formation of different regioisomers or isomers as well as any possible changes in stereochemistry – knowledge which allows chemists to design synthetic routes specifically targeting their desired products.
Mechanistic Insight: Hydride and methyl shifts provide valuable insight into the mechanisms underlying organic reactions. By studying these shifts, chemists gain a better understanding of how reactions occur, including carbocation intermediates’ roles and stabilization techniques. With this knowledge at their disposal, new reaction pathways, increased reaction efficiency, and expanded synthetic toolbox can be created.
Controlling Selectivity: Regioselectivity control is a core part of synthetic organic chemistry. Hydride and methyl shifts can significantly change regioselectivity by changing where migratory groups migrate; understanding their selectivity factors allows chemists to fine-tune reactions and direct formation of desired products more precisely.
Synthetic Strategy: Understanding hydride and methyl shifts allows chemists to further their synthetic strategies. By strategically taking advantage of these shifts, they can access different reaction pathways for synthesizing complex molecules – expanding their synthetic toolbox and helping build novel molecular architectures.
Understanding Hydride/Methyl Shifts Can Have Positive Implications in Medicinal Chemistry and Drug Discovery: Understanding hydride/methyl shifts can have substantial ramifications for medicinal chemistry and drug discovery. Slight variations in functional group placement within molecules can have profound ramifications on their biological activity, so by recognizing hydride or methyl shifts within drug molecules or lead compounds medicinal chemists can optimize them further to enhance potency, selectivity or pharmacokinetic properties of their molecules/lead compounds for maximum performance.
Understanding the distinctions between hydride and methyl shifts is vital to accurately predicting reaction outcomes, gaining mechanistic insights, controlling selectivity, developing synthetic strategies, optimizing molecular structures in organic chemistry fields across many applications and optimizing molecular structures for various molecular structures in various applications. Knowledge of this field also allows chemists to design efficient synthetic routes while better comprehending organic reactions for scientific research and development purposes.
Carbocation stability and rearrangement
Carbocation stability and rearrangement are closely intertwined in organic chemistry. Carbocations, positively charged carbon species that lack electron-deficient p orbitals, are highly reactive substances capable of participating in various chemical transformations; yet due to their unstable nature they frequently undergo rearrangements that form more stable intermediates or products.
Carbocation Stability:
Carbocation stability refers to the relative energy levels and reactivity of different carbocation species, most notably their electron delocalization or presence of electron-donating groups nearby the positively charged carbon atom.
As more electron-donating groups appear near its positively charged carbon core, its stability increases. The greater this delocalization or presence, the more stable a carbocation becomes.
Carbocations stability follows this order: tertiary > secondary > primary > methyl. Tertiary carbocations is more stable due to having more neighboring carbon atoms that donate electron density, leading to greater electron delocalization and stability; on the contrary, primary and methyl carbocations lack electron-donating groups, making them among the least stable species.
Carbocation Rearrangement:
A carbocation rearrangement refers to the process by which alkyl groups or hydrogen atoms migrate within an intermediate carbocation, in order to remove its positive charge and create more stable carbocations products or intermediate states. Carbocation rearrangements occur with the goal of creating more secure intermediate states or transition states as their driving force.
Hydride shifts and alkyl shifts are two popular carbocation rearrangements. Hydride shifts involve moving hydride ions (H-) from nearby carbon atoms onto an adjacent carbon atom of the carbocation to make it more stable, while alkyl shifts involve moving an alkyl group from one carbon atom onto another, creating a rearrangement.
Carbocation rearrangements have the ability to dramatically change the regioselectivity and stereochemistry of organic reactions, altering carbon connectivity to produce isomers or regioisomers that may result in isomer formation. Therefore, understanding and anticipating carbocation rearrangements is integral in designing synthetic routes and controlling outcomes of organic reactions.
Carbocation stability and rearrangement are fundamental concepts in organic chemistry. Carbocations are stabilized through electron delocalization and electron-donating groups nearby; however, due to their inherent instability they often undergo rearrangements that form more stable intermediates; such rearrangements may impact regioselectivity or stereochemistry during reactions, making these considerations essential in synthetic strategies and reaction design.
Impact on product formation and stereochemistry
Carbocation stability and rearrangement play an enormously influential role in organic reactions, affecting product formation and stereochemistry in significant ways. Let’s investigate these aspects more in-depth:
Product Formation:
Carbocation Stability: More stable carbocations can be created through resonance or the presence of electron-donating groups nearby, which affects reaction outcomes by determining which major product forms from reactions that involve carbocation intermediates – for instance addition and substitution reactions often produce products containing only those carbocations intermediates that form more stable carbocations intermediates.
Carbocation Rearrangement: Rearrangements can result in the formation of various isomers or regioisomers, due to migration of alkyl groups or hydrogen atoms within a carbocation intermediate that alters carbon connectivity, altering product distribution. Depending on the nature of this rearrangement, different positional isomers or constitutional isomers may form, altering overall product profile.
Stereochemistry:
Carbocation Stability: Carbocation stability does not typically influence the stereochemistry of reactions; creating more stable carbocation intermediates does not create or modify new chiral centers, or alter existing stereocenters; hence the stereochemistry is usually determined by other factors like reactant stereochemistry, reaction conditions or presence of chiral catalysts.
Carbocation Rearrangement: Carbocation rearrangements can significantly change the stereochemistry of reactions by shifting relative positions of substituents. When an alkyl group migrates during a carbocation rearrangement, this could alter its stereochemistry; leading to diastereomers or enantiomers being formed as a result.
Carbocation rearrangements follow the principles of stereochemistry, with groups migrating with either retention or inversion depending on reaction conditions and mechanisms involved. Therefore, understanding stereochemical implications is vital in order to predict and control organic reactions accurately.
Carbocation stability determines which products form in reactions involving carbocation intermediates, while rearrangements can lead to isomers or regioisomers forming, altering the product profile and potentially altering stereochemistry of reactions.
While stability usually does not directly alter stereochemistry directly, rearrangements may alter spatial arrangements of substituents leading to stereochemical consequences – an understanding of these effects is critical when designing reactions and controlling organic transformations.
Adjacent functional groups or substituents
Functional groups or substituents of organic molecules that are adjacent can have a dramatic effect on their reactivity and selectivity of reactions, from intermediate stability and stereochemistry effects to regioselectivity and regioselectivity of reactions. Let’s investigate their influence more in depth:
Stability of Intermediates:
Electron-donating or electron-withdrawing groups nearby can influence the stability of reaction intermediates such as carbocations, radicals and anions. Donor groups can stabilize positive charges or radicals by providing electron density through resonance or inductive effects while withdrawers destabilize intermediates by withdrawing electron density through resonance or inductive effects – ultimately impacting reaction rates and overall outcomes of reactions.
Regioselectivity: Adjacent functional groups or substitutes can have an effect on the regioselectivity of reactions, which refers to their preference to occur at particular sites within molecules. Specific functional groups can direct reactions toward one location through electronic or steric effects – for instance in electrophilic aromatic substitution reactions electron-donating or withdrawing groups on aromatic rings can influence where substitution takes place.
Stereochemistry: Neighboring functional groups or substituents can have an enormous effect on the stereochemistry of reactions. Their proximity can create steric hindrance that results in preferential formation of certain stereoisomers, or neighboring functional groups can participate in stereospecific reactions whereby their reactant’s original stereochemistry remains preserved in its product – for instance in neighboring group participation (NGP) reactions where its stereochemistry is determined by that of adjacent functional groups.
Reactive Intermediates: Adjacent functional groups or substituents can alter the reactivity of reactive intermediates. For instance, adjacent electron-donating groups may increase nucleophilicity or stability of nucleophiles while adjacent electron-withdrawing groups can decrease nucleophilicity and enhance electrophilicity – these effects having an impactful on reaction rates and selectivity of reactive sites within molecules.
Understanding the effects of adjacent functional groups or substituents is vital for designing synthetic routes, predicting reaction outcomes and controlling stereochemistry and regioselectivity of reactions. By considering these effects, chemists can strategically modify molecular structures to achieve desired reactivity and selectivity in organic transformations.
Influence on reaction rate and product distribution
Functional groups or substituents present on organic molecules can have an enormous impact on reaction rates and product distribution, so let’s explore their role:
Reaction Rates:
Functional groups or substituents located nearby can have an effect on reaction rates by altering electronic and steric properties of reactants, leading to different rates in different scenarios. Here are some scenarios:
Electronic Effects: Electron-donating or electron-withdrawing groups can modify electron density within molecules, altering nucleophilicity or electrophilicity of reactive species and hastening reactions with electrophiles or nucleophiles, respectively. Electron-donating groups can increase nucleophilicity for faster reactions with electrophiles while electron-withdrawing groups decrease nucleophilicity or increase electrophilicity to speed reactions with nucleophiles more quickly.
Steric Effects: Bulky substituents can produce steric hindrance that reduces access to reactive sites, slowing the reaction rate. Small substituents or functional groups with minimal steric hindrance allow more efficient collisions and faster reaction rates.
Adjacent functional groups or substitutes can have an impactful effect on product distribution in an interaction, including:
Regioselectivity: Regioselectivity refers to the preference for one regioisomer over others in chemical reactions. Functional groups or substituents nearby can influence this preference through electronic or steric effects; such effects could direct reactions at specific sites within molecules, leading to specific regioisomeric products.
Stereoselectivity: Stereoselectivity refers to the selective formation of one stereoisomer over others. Adjacent functional groups or substituents may create steric interactions or provide stereoelectronic effects which favor selective formation of certain stereoisomers; this may lead to diastereoselective or enantioselective reactions in which certain stereoisomeric products are preferred over others.
Neighboring Group Participation (NGP): Neighboring group participation (NGP) refers to an effect in which adjacent functional groups or substituents participate in a chemical reaction and alter its stereochemistry, often leading to stereospecific transformations whereby the stereochemistry of the reactant remains preserved in its product due to NGP reactions involving adjacent groups.
Consideration of adjacent functional groups or substituents is vital in order to accurately predict reaction rates and analyze product distribution in organic transformations. Understanding their effects allows chemists to optimize reaction conditions, control regioselectivity, stereoselectivity and design synthetic routes with desired product profiles.
Stereochemical outcomes and implications
Substituents in organic molecules can have profound impacts on the stereochemical outcomes of reactions, with adjacent functional groups having various influences over product stereoisomericity through various mechanisms, leading to different stereoisomeric forms. Let’s examine some of these implications:
Stereoselectivity:
Functional groups or substituents that interact can produce steric hindrance or stereoelectronic effects that lead to preferential formation of certain stereoisomers; this phenomenon is known as stereoselectivity.
Their presence may alter reagent approach rates, bond formation orientation or intermediate stability which then favor one stereoisomer over the others, providing controlled production of specific stereoisomers with desired properties. Stereoselective reactions are highly useful tools in organic synthesis as they enable controlled synthesis of specific stereoisomers with desired properties resulting in their controlled production of certain stereoisomers with desired properties resulting in stereoselective reactions producing specific stereoisomers with desired properties resulting in controlled production of specific stereoisomers with desired properties for controlled production of desired properties desired in controlled production processes that lead to preferential formation over another.
These reactions make use of these groups present to influence approach of reagents to influence approach of reagents approaching from being present can influence approach from approaching reagents approaching from being present thus leading to influence approach of reagents being present resulting in selective formation over another.
These reactions make use of selective organic synthesis as they allow controlled production of desired stereoisomers with desired properties allowing controlled synthesis processes with desired characteristics being produced as desired resulting in controlled reactions with desirable properties being produced thereby.
These selective reactions make them extremely valuable tools of organic synthesis because their controlled creation allows controlled production of specific stereoisomers with desired properties being produced through bond formation orientation orientation bond formation orientation bond formation or stability intermediate formation and stable intermediate formation thus leading to formation or stability of intermediates leading to formation leading to one stereoisomer over another which in organic allowing controlled synthetic chemicals enabling controll synthesis with desired properties than non selective reactions allow controlled allowing controlled allowing controlled allowing controlled production with specific properties from one over another which results.
This allows controlled synthetic organic resulting in controlled organic synthelective reactions which allow controlled synthetic process enabling targeted for controlled production with desired properties being produced for desired properties of desired allowing targeted via controlled synthetic route than others would otherwise would allow controlled synthetic route or stabilization processes, leading to selective reactions as desired intermediate formation through controlled production process to emerges while synthe/isomer synthesis using organic synthe/other then results.
Steric Effects: Bulky substitutents nearby can create steric hindrance that affects bond formation during reactions. This steric hindrance may influence the stereochemistry of products; in reactions involving chiral molecules, this interaction between adjacent groups may result in diastereoselectivity – in which specific diastereomers form at higher yields; additionally, using chiral auxiliary groups or catalysts may further enhance stereoselectivity by providing extra stereocontrol elements.
Neighboring Group Participation (NGP) is a stereochemical phenomenon in which nearby functional groups or substituents participate in a reaction, altering its stereochemistry. NGP may occur through intramolecular interactions whereby adjacent functional groups or substituents participate in bond formation or cleavage, with this participation inducing specific stereochemical relationships between reactant and product molecules that influence stereochemical aspects of a reaction.
Chirality Transfer: mes The presence of multiple chiral centers or groups can result in the transfer of chirality from one part of a molecule to another through reactions involving neighboring groups or rearrangements, keeping or inverting stereochemistry while maintaining or inverting it respectively in products. Chirality transfer plays a significant role in asymmetric synthesis as it allows the introduction or preservation of stereochemistry even in complex molecules.
Understanding the effects of adjacent functional groups or substituents on stereochemistry is critical in designing synthetic routes and controlling formation of specific stereoisomers. Chemists can leverage these effects to achieve desired stereochemical outcomes, produce enantiomerically pure compounds or access stereoisomeric libraries for biological screening purposes. Furthermore, understanding stereochemistry enables interpretation of biological activities, understanding structure-activity relationships and designing chiral drugs or catalysts.
Conclusion
Acknowledging hydride and methyl shifts is fundamental in organic chemistry. Hydride shifts involve migrations of H- groups from one carbon atom to another; on the other hand, methyl shifts involve movement of CH3 groups between carbon atoms; these occur frequently during carbocation reactions that play an integral part in determining their outcomes.
Scientists use the distinction between hydride and methyl shifts to more accurately predict regioselectivity and stereochemistry of reactions. Hydride shifts alter the position of migrating hydrides, impacting on their regioselectivity of reaction; on the other hand, methyl shifts have an impactful impact on regioselectivity but usually don’t change stereochemistry significantly.