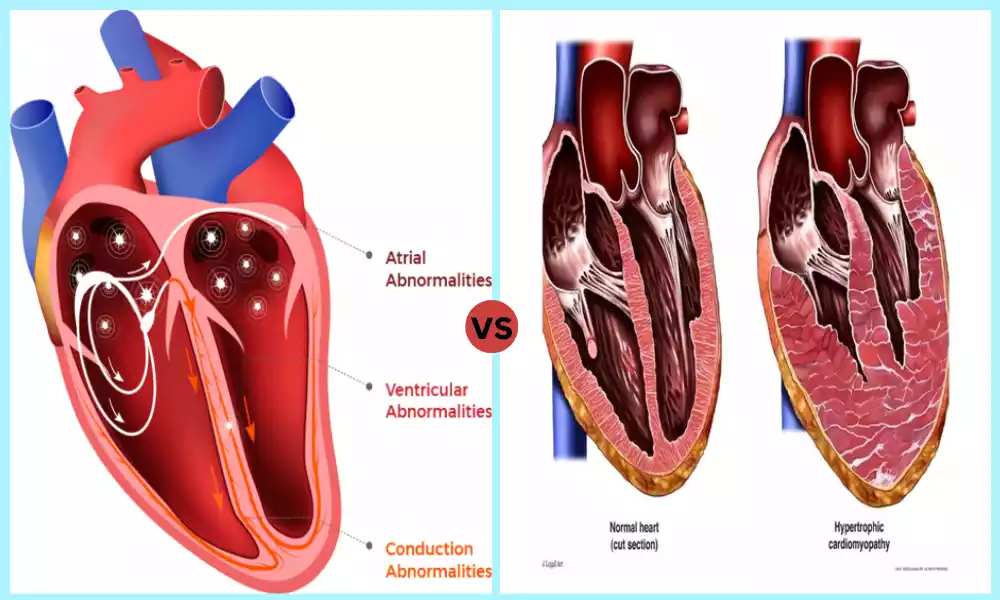
Introduction of Dilated Cardiomyopathy and Hypertrophic Cardiomyopathy
Cardiomyopathy refers to a group of diseases that impact the structure and function of heart muscle. Of the various forms of Cardiomyopathy, dilated Cardiomyopathy (DCM) and hypertrophic Cardiomyopathy (HCM) stand out due to Significant differences in their Characteristics, causes, clinical Presentations, diagnostic approaches, treatment approaches and overall patient outcomes. Recognizing DCM/HCM differences is paramount to accurate diagnosis, effective management strategies and improved patient outcomes.
Dilated Cardiomyopathy (DCM) is characterized by the Enlargement and weakening of the left ventricle, the heart muscle Responsible for pumping oxygenated blood to other parts of the body. Dilation and thinning lead to reduced Contractility and compromised pumping function that leads to symptoms like fatigue, shortness of breath, fluid retention and eventually heart failure. Causes include genetic mutations, viral infections, toxins or systemic diseases as well as any underlying factors.
Hypertrophic Cardiomyopathy (HCM), on the other hand, refers to an Abnormal thickening of heart muscle in Particular in the left Ventricle. This hypertrophy may obstruct normal blood flow and impair its pumping function resulting in chest pain, shortness of breath, dizziness or fainting as symptoms; oftentimes caused by genetic mutations affecting proteins involved with cardiac muscle contraction.
DCM and HCM are two forms of primary cardiomyopathy, yet they differ significantly in terms of cardiac structure, genetic factors, clinical presentation, diagnostic criteria, treatment options and long-term outcomes. This content outline will explore these differences extensively by exploring each condition’s unique features while emphasizing the significance of timely diagnosis and appropriate management for individuals affected by either DCM or HCM.
Definition of Dilated Cardiomyopathy
Dilated Cardiomyopathy (DCM) is a primary cardiomyopathy characterized by an enlarged left ventricle of the heart with reduced pumping function – its main pump chamber. DCM occurs when the heart muscle becomes thin and stretched, decreasing contractility and hindering its ability to pump blood efficiently.
Dilation and Weakening of the heart muscle can have severe Detrimental effects on its ability to Circulate blood Throughout the body Efficiently, leading to fatigue, Shortness of breath, fluid Retention and Eventually heart Failure.
DCM can affect people of all ages and can be caused by various Factors, including genetic Mutations, viral infections, exposure to toxins or Medications as well as Systemic diseases like diabetes or Thyroid Disorders. Early diagnosis and effective management are critical to slowing its progress while improving quality of life for those affected.
Definition of Hypertrophic Cardiomyopathy
Hypertrophic Cardio Myopathy (HCM) is a primary Cardiomyopathy characterized by Abnormal thickening (hypertrophy) of heart Muscles, particularly in the left Ventricle. HCM occurs when heart muscle thickens beyond normal, Interfering with its ability to pump blood Efficiently. Additionally, this Thickening may alter heart structure by narrowing heart chambers or stiffening of ventricular walls.
HCM is typically caused by genetic mutations that alter proteins responsible for contracting heart muscle cells. HCM can result in abnormal thickening and structural changes to the heart that interfere with normal blood flow, leading to symptoms like chest pain, shortness of breath, dizziness, fainting and palpitations.
HCM may also increase your risk for abnormal heart rhythms as well as sudden cardiac arrest in rare instances. HCM can affect people of any age, although it often appears during adolescence or young adulthood.
Treatments for HCM aim to alleviate symptoms, prevent complications, and lower sudden cardiac events by taking medications, making lifestyle modifications, or in some cases using surgical procedures like septal myectomy or alcohol septal ablation. Regular monitoring and follow-up care is essential in managing HCM effectively.
Comparison Table of Dilated Cardiomyopathy and Hypertrophic Cardiomyopathy
Sure! Here’s a comparison table outlining the key differences between Dilated Cardiomyopathy (DCM) and Hypertrophic Cardiomyopathy (HCM):
| Aspect | Dilated Cardiomyopathy (DCM) | Hypertrophic Cardiomyopathy (HCM) |
|---|---|---|
| Definition | Enlargement and weakening of the left ventricle | Abnormal thickening of the heart muscle, particularly the left ventricle |
| Cardiac Structure | Dilated and thin heart muscle | Hypertrophied (thickened) heart muscle |
| Genetic Factors | Genetic mutations can be present | Genetic mutations are the primary cause |
| Etiology | Genetic mutations, viral infections, toxins, underlying systemic diseases | Primarily genetic mutations affecting proteins involved in cardiac muscle contraction |
| Clinical Presentation | Fatigue, shortness of breath, fluid retention, heart failure symptoms | Chest pain, shortness of breath, dizziness, fainting |
| Diagnostic Approach | Medical history, physical examination, echocardiography, electrocardiogram, cardiac MRI | Medical history, physical examination, echocardiography, electrocardiogram, genetic testing |
| Treatment Options | Medications, device therapy (implantable cardioverter-defibrillator), cardiac transplantation | Medications, septal myectomy, alcohol septal ablation |
| Prognosis | Varies depending on the underlying cause and response to treatment | Varies depending on the severity and presence of complications |
| Long-term Management | Regular monitoring, lifestyle modifications, medication adherence | Regular monitoring, lifestyle modifications, medication adherence, potential surgical interventions |
It’s important to note that while this table highlights the primary differences between DCM and HCM, individual cases may exhibit variations in symptoms, progression, and treatment approaches. Consulting with a healthcare professional is crucial for accurate diagnosis, personalized treatment plans, and ongoing management.
Importance of understanding the differences between DCM and HCM
Understanding the differences between Dilated Cardiomyopathy (DCM) and Hypertrophic Cardiomyopathy (HCM) is of great significance for several reasons, including:
Accurate Diagnosis: DCM and HCM each present with unique symptoms and diagnostic criteria, making identification and differentiation between them essential to healthcare professionals in determining an accurate diagnosis, leading to timely initiation of appropriate treatments strategies and timely patient recovery.
Healthcare providers need to know these differences to accurately differentiate them when making diagnoses. Understanding them allows healthcare professionals to accurately distinguish them and perform appropriate diagnostic tests accordingly for timely initiation of appropriate therapy plans.
Treatment Selection: DCM and HCM may require different approaches for their management. DCM often relies on medications to manage heart failure symptoms; in severe cases cardiac transplantation may be required.
For HCM sufferers, medications may relieve symptoms while surgical interventions like septal myectomy or alcohol septal ablation could provide additional support. Understanding these differences helps healthcare providers select effective options tailored specifically to each patient.
Prognostic Assessment: Prognostic outcomes vary between DCM and HCM. Gaining an in-depth knowledge of these differences enables healthcare providers to provide accurate prognostic information to patients and their families, setting appropriate expectations, establishing personalized management plans, and leading discussions regarding possible complications or disease progression.
Genetic Counseling and Testing: Genetic factors play a major role in both DCM and HCM; however, their respective genetic mutations and inheritance patterns differ substantially between them.
Understanding these distinctions is critical when providing genetic counseling or genetic testing; healthcare professionals need this knowledge in order to provide accurate advice to patients as well as their families regarding risk of familial transmission or potential implications on other family members.
Research and Advancements: Recognizing the differences between DCM and HCM allows researchers to focus their research efforts more precisely for each condition. As researchers focus on each cardiomyopathy’s unique characteristics, advancements can occur faster in understanding its underlying mechanisms, genetic influences, treatment approaches, and patient outcomes.
By understanding more effectively these differences amongst cardiomyopathies can result in more effective therapies with improved patient outcomes.
Understanding the differences between DCM and HCM allows for accurate diagnosis, appropriate treatment selection, prognostic assessment, genetic counseling and advancements in research. In turn, this knowledge improves patient care, outcomes and contributes to new strategies for managing cardiomyopathies.
Similarities Between DCM and HCM
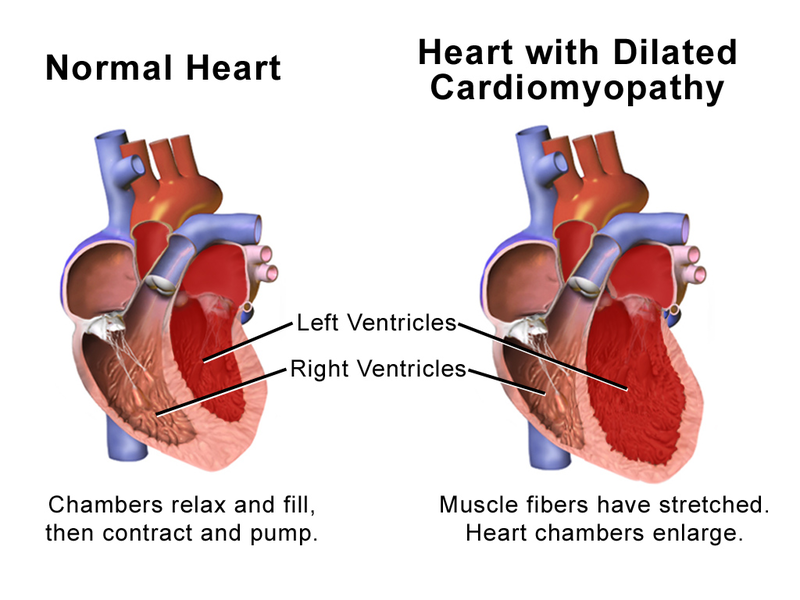
Both Dilated Cardiomyopathy and Hypertrophic Cardiomyopathy share certain Distinctive characteristics, but there can also be similarities. Some Examples include:
Primary Cardiomyopathies : DCM and HCM are both primary cardiomyopathies. This means that they directly affect the heart muscle’s structure and function, rather than secondary to other causes like hypertension or coronary arterial disease.
Progressive Diseases: Both DCM and HCM have the potential for progression over time. Although rates of progression can differ among individuals, both conditions could worsen over time, leading to symptoms and complications for those living with either disease. Therefore, regular monitoring and appropriate management is vital to slow disease progression and ensure better outcomes.
Risk of Arrhythmias: Both DCM and HCM Increase the risk of Arrhythmias, or abnormal heart Rhythms, such as Palpitations to more serious Arrhythmias such as atrial Fibrillation or ventricular Arrhythmias. Close monitoring and, if necessary, appropriate interventions are essential to manage and treat arrhythmias in both conditions.
Lifestyle Modifications Are Vital: Lifestyle modifications play a vital role in managing both DCM and HCM. Such lifestyle modifications may include regular physical activity within recommended limits, eating heart-healthy food, managing weight, refraining from alcohol and tobacco, as well as stress reduction techniques.
By adopting such changes into daily life, individuals with either condition can help improve symptoms, overall heart health, quality of life and ultimately life expectancy.
Need for Regular Monitoring and Follow-up: Both DCM and HCM require ongoing monitoring with healthcare providers, including regular evaluation of cardiac function, symptoms, medication adjustments and possible screening for complications. Regular follow-up ensures that these conditions are effectively managed with appropriate interventions implemented when needed.
While understanding the differences between DCM and HCM are important, understanding their shared characteristics helps identify common management strategies as well as emphasizes the necessity of comprehensive care and follow-up for those affected by cardiomyopathies.
Medical history and physical examination
Medical history review and physical Examination are integral parts of Diagnosing Dilated Cardiomyopathy (DCM) and Hypertrophic Cardiomyopathy (HCM), providing Healthcare providers with invaluable Information, while serving as initial steps in Evaluating individuals Suspected of Cardiomyopathy.
Here is an outline of their role in Diagnosing DCM and HCM:
Medical History: Symptom Analysis: Patients will be asked about any presenting symptoms such as fatigue, shortness of breath, chest pain, palpitations and exercise intolerance that they are experiencing. Their nature, frequency and duration provide insights into any underlying conditions they might have.
Past Medical History: Gathered information includes prior heart-related conditions such as coronary artery disease, heart attacks, hypertension or congenital heart disease. Furthermore, any history of systemic diseases, viral infections or toxic exposures or family history of cardiomyopathy must also be assessed as potential sources of it.
Medication and Allergies: Healthcare providers assess any medications taken by their patient, particularly cardiac-related ones, which could contribute to or exacerbate cardiomyopathy. They will also note any allergies related to medications taken.
Family History: Examining family histories related to cardiomyopathy, sudden cardiac death or other relevant cardiac conditions is of vital importance when investigating DCM and HCM as genetic factors play a significant role. Accurate information regarding affected relatives such as their age of diagnosis or genetic testing results will aid risk assessments and genetic counseling services.
Physical Exam: (General Appearance and Vital Signs). Vital signs including blood pressure, heart rate and respiratory rate will be measured during this examination.
Auscultation: Healthcare providers use a stethoscope to listen to a patient’s heart sounds and pay special attention to any abnormal sounds such as murmurs or gallops that could indicate structural abnormalities within their cardiovascular system.
Pulmonary Examination: Healthcare providers conduct a pulmonary exam on the patient to detect any evidence of fluid accumulation or congestion in their lungs that might indicate heart failure.
Peripheral Edema: Swelling in the legs, ankles or feet should be monitored as it could indicate fluid retention due to heart dysfunction.
Chest Exam: A chest examination includes looking for signs of irregular cardiac impulses or thrills, which could indicate either an enlarged heart or irregular heart rhythm.
Medical history and physical examination findings provide essential initial clues in diagnosing DCM or HCM.
Healthcare providers use these assessments to decide whether additional diagnostic tests, such as echocardiography, electrocardiography or genetic testing are needed in order to confirm cardiomyopathy and formulate appropriate management strategies.
Prognosis and long-term management
Prognosis and long-term management strategies for Dilated Cardiomyopathy (DCM) and Hypertrophic Cardiomyopathy (HCM) are crucial in order to optimize patient outcomes and quality of life.
Below is an overview of some prognostic/long-term management approaches available for each disease:
Prognosis for Dilated Cardiomyopathy (DCM):
Prognosis can vary significantly depending on a variety of factors, including its cause, symptoms severity, treatment response and any complications present. DCM poses risks including progressive heart failure, arrhythmias and sudden cardiac death.
Individuals may experience disease stabilization or improvement with proper management; others may progress more rapidly. for Hypertrophic Cardiomyopathy (HCM).
Prognosis for HCM depends on factors like its degree, symptoms and risk for sudden cardiac death. Although most individuals with HCM lead relatively stable clinical courses, some may experience disease progression or develop complications.
Identification of high-risk features such as severe hypertrophy, family history of sudden cardiac death or certain abnormal cardiac findings can help stratify risks and guide management decisions.
Long-Term Management:
Medications may help manage conditions over the longer term.
Both DCM and HCM may require medications to manage symptoms, regulate heart rhythm, and maximize cardiac health.
Medication typically used includes beta-blockers, calcium channel blockers, angiotensin-converting enzyme (ACE) inhibitors, angiotensin receptor blockers (ARBs), diuretics and antiarrhythmic drugs. Individualized treatment plans may also include lifestyle modifications.
Lifestyle changes are an integral component of successful DCM and HCM management. Patients should adopt a heart-healthy diet low in sodium and saturated fats, engage in regular physical activity within recommended limits, limit alcohol consumption to the recommended limit, quit smoking altogether and effectively manage stress.
Weight Management and Regular Monitoring/Follow-up: These activities should also be prioritized. In particular, blood pressure monitoring is often stressed. When applicable, glucose levels (if applicable) must also be regularly tracked for any health complications or changes. Specifically: Regular Follow-up of B.P and/or blood glucose Levels is encouraged as a priority.
Ongoing monitoring is crucial to assess disease progression, optimize medication regimens and identify any complications.
Frequent visits with healthcare providers, including cardiologists, should be scheduled in order to monitor symptoms, perform cardiac imaging (such as echocardiography), and conduct electrocardiograms.
Additional tests such as cardiac MRI, exercise stress testing or genetic testing may also be advised for further risk assessment and intervention purposes. (Risk stratification and interventions:)
Risk stratification for complications such as sudden cardiac death or thromboembolism allows healthcare providers to identify individuals who could benefit from particular interventions.
High-risk individuals at increased risk for HCM should consider implantable cardioverter defibrillator (ICD) placement or septal reduction therapies like septal myectomy or alcohol septal ablation to lower their heart rhythm risk.
When medical therapies do not effectively manage DCM or HCM, cardiac transplantation or the installation of a ventricular assist device (VAD) may become necessary. As part of such procedures, genetic counseling and family screening must also be considered.
Genetic counseling and family screenings are integral components of effective management for both DCM and HCM. Identification of genetic mutations may aid screening efforts while providing risk assessments for any affected relatives.
Long-term management in DCM and HCM seeks to optimize cardiac function, prevent disease progression, reduce symptoms, and mitigate complications. Regular monitoring, appropriate medication regimens, lifestyle modifications, risk stratification analysis and interventions all play a part in providing comprehensive care to individuals diagnosed with cardiomyopathies such as DCM or HCM.
Conclusion
Dilated Cardiomyopathy (DCM) and Hypertrophic Cardiomyopathy (HCM) are two distinct primary Cardiomyopathies with Significant variations in Characteristics, causes, clinical Presentations, diagnostic Approaches and treatment Strategies that must be Understood accurately for accurate diagnosis, Appropriate management and improved patient outcomes. Understanding these variations is critical in accurate diagnosis, management and improving patient outcomes.
DCM is a condition in which the left ventricle becomes enlarged and weaker, leading to reduced contractility and pumping function, often as the result of genetic mutations, viral infections, toxins or systemic diseases.